A Genetic Reason for Breathlessness
Majdi Al Nabulsi MD, Pulmonary Critical Care Fellow
Zeina AlMajthoub, Pulmonary Critical Care Research Associate
Kathryn A. Radigan MD, Pulmonary Critical Care Attending
John H. Stroger Hospital of Cook County
Chicago, Illinois
Case
A 48 year-old male with past medical history significant for hepatitis C and cirrhosis presented to the hospital for decompensated liver disease. Pulmonary team was consulted for an abnormal CT scan of the chest. The patient admitted to dyspnea for months which was associated with audible wheezing. He smoked around 6 cigarettes a day for 21 years. He denied significant occupational exposures.
Images
Question
What is the most appropriate test that should be requested for this patient?
- Sweat Chloride Test
- Sputum AFB Smear & Culture
- Serum Alpha-1 Antitrypsin Level
- Serum Immunoglobulins Levels
- ANA Titer
C: Serum Alpha-1 Antitrypsin Level
Discussion
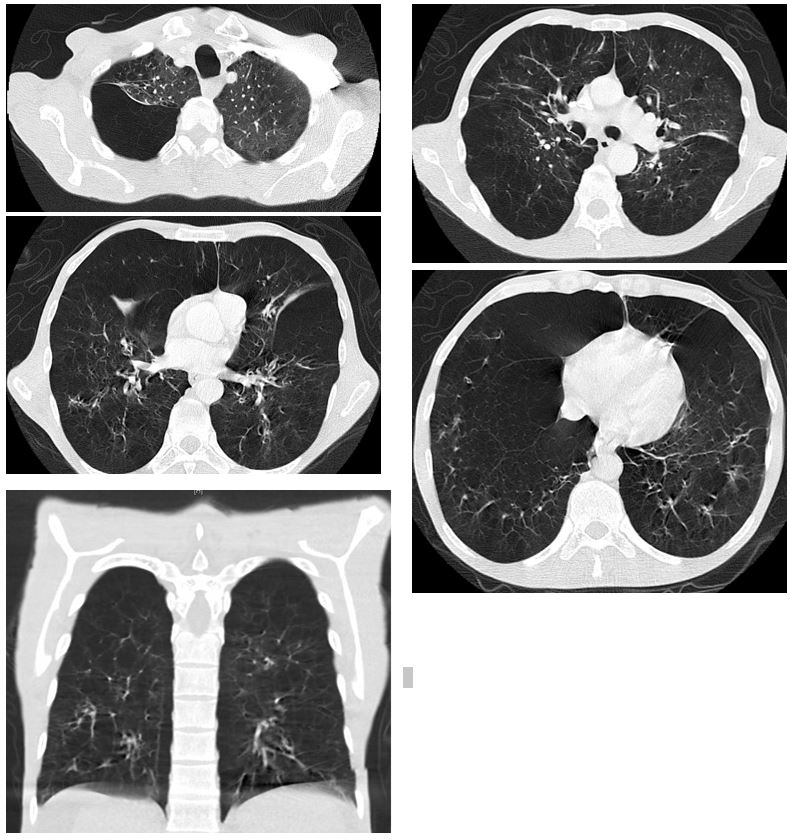
This middle age man has extensive panlobular emphysema with basilar predominance on imaging along with liver cirrhosis. These two conditions together raise concern for alpha-1 antitrypsin [A1AT] deficiency. His serum A1AT level was very low at 19 mg/dL. His genotype was positive for two copies of the Z allele (ZZ). His pulmonary function test showed FVC 58%, FEV1 23%, and FEV1/FVC ratio of 31% indicating a very severe obstructive ventilatory defect.
Although a highly underrecognized genetic disorder, A1AT deficiency is relatively common. In most patients, the diagnosis is delayed for at least a couple of years after symptoms onset. (1) The inheritance is autosomal codominant with approximately 150 alleles [normal and defective] that have been identified thus far leading to different combinations in these gene loci. The phenotype and severity depend on the level of serum A1AT produced by these alleles. Individuals with low serum levels should be further tested with isoelectric focusing or genotyping to confirm the diagnosis. (2,3)
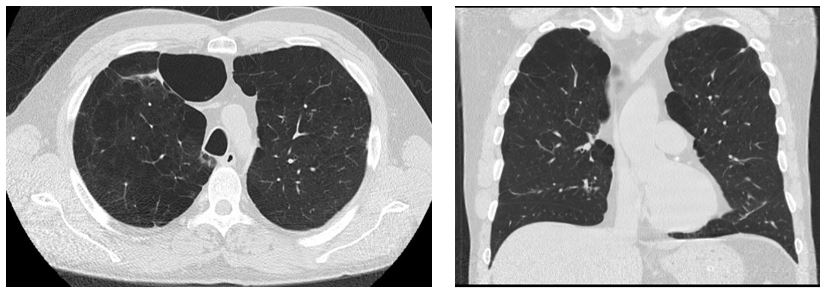
Clinical manifestations include lung disease (emphysema and bronchiectasis), liver disease (chronic hepatitis, cirrhosis, and hepatocellular carcinoma), and rarely skin disease including necrotizing panniculitis along with psoriasis and urticaria. Emphysema here has early onset (typically the 4th or 5th decades) with panlobular pattern that is characterized by disproportionate emphysematous involvement of the lung bases and destruction of the entire alveolus (secondary pulmonary lobule) uniformly. (2) This is different from the other more common pattern, centrilobular emphysema, which is localized to the proximal respiratory bronchioles with normal surrounding lung parenchyma, focal destruction, untouched distal alveolar ducts and sacs, and predominantly found in the upper lung zones (See the picture to compare). (4) This latter form occurs with long-standing tobacco smoking and dust inhalation. Although both ATS and WHO guidelines recommend screening for A1AT deficiency in all COPD patients, its importance is stressed in patients with early onset emphysema, bronchiectasis, liver disease without a clear etiology, unexplained panniculitis, and in patients with anti-proteinase-3 vasculitis. (2,5) Interestingly, some genotypes spare the liver and cause lung disease only.
Centrilobular Emphysema pattern (For comparison)
Besides conventional COPD management, smoking cessation and pulmonary rehabilitation, treatment also involves infusion of pooled human alpha-1 proteinase inhibitor. This treatment is effective in reducing frequency of exacerbations, FEV1 decline, and overall mortality in carefully selected patients. (2,6) However, the high cost of such treatment remains a challenge that is unaffordable by many patients.
References
-
Silverman EK, Miletich JP, Pierce JA, et al. Alpha-1-antitrypsin deficiency. High prevalence in the St. Louis area determined by direct population screening. Am Rev Respir Dis 1989; 140(4): 961-6.
-
Stoller JK and Aboussouan LS. A Review of α1-Antitrypsin Deficiency. Am J Respir Crit Care Med 2012; 185(3): 246-59.
-
American Thoracic Society/European Respiratory Society Statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168(7): 818–900.
-
ATS Committee on Diagnostic Standards for Nontuberculous Respiratory Diseases, American Thoracic Society. Definitions and classification of chronic bronchitis, asthma, and pulmonary emphysema. Am Rev Respir Dis 1962; 85: 762–9.
-
Alpha 1-antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ 1997; 75(5): 397-415.
-
Marciniuk DD, Hernandez P, Balter M, et al. Alpha-1 antitrypsin deficiency targeted testing and augmentation therapy: a Canadian Thoracic Society clinical practice guideline. Can Respir J 2012; 19(2): 109-16.


