Alphabet Soup: Differentiating Interstitial Lung Diseases
Authors: Daniel G. Dunlap, MD1; Jared Chiarchiaro, MD1
Affiliations: 1Division of Pulmonary, Allergy, and Critical Care Medicine, University of Pittsburgh Medical Center, Pittsburgh, PA
Case
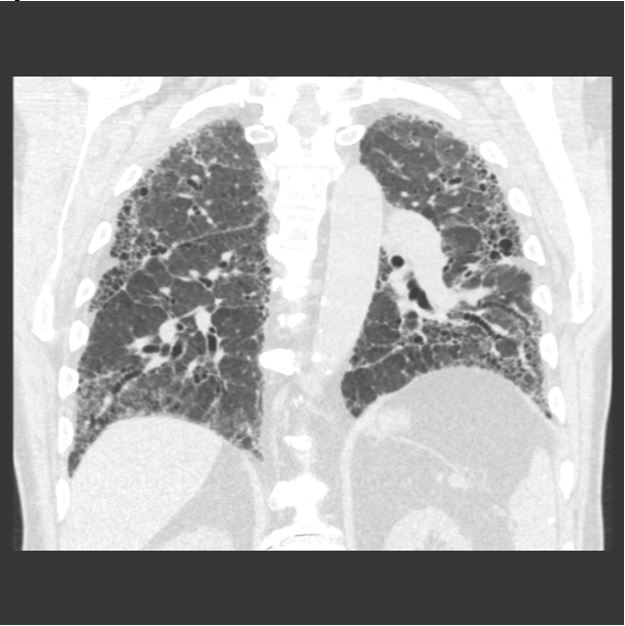
A previously healthy, 67-year-old male presents with dyspnea on exertion and an abnormal chest radiograph. The patient reports 1.5 years of progressively worsening dyspnea on exertion, now requiring supplemental oxygen with activity. CT chest demonstrates the following (Figure 1A and IB):
Figure 1A:
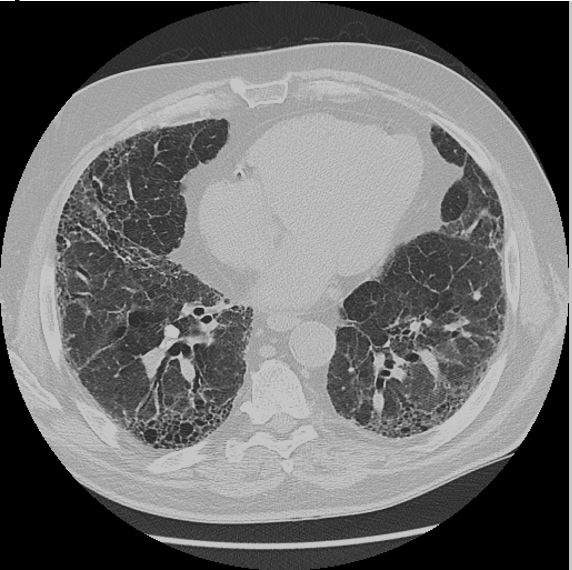
Figure 1B:
Question
What is the clinician’s next step in evaluation?
- Bronchoscopy with transbronchial biopsies
- VATS biopsy
- Obtain further history
- Prednisone burst
- Referral for lung transplantation
Answer: C. Obtain further history
Discussion
Obtaining a complete social and occupational history is critical when evaluating patients with interstitial lung disease (ILD). This patient’s CT chest demonstrates traction bronchiectasis and honeycombing, consistent with a usual interstitial pneumonia (UIP) pattern of fibrosis. UIP is the typical histopathologic finding in idiopathic pulmonary fibrosis (IPF), but may also be seen in a myriad of other end-stage pulmonary diseases. This patient lives on a farm and frequently works in a silo. Furthermore, his symptoms flare when working on the farm and improve when he limits his exposures. Thus, he was diagnosed with hypersensitivity pneumonitis (HP).
Hypersensitivity pneumonitis, also known as extrinsic allergic alveolitis, refers to inflammation of the alveoli, bronchioli, and alveolar interstitium because of a delayed allergic reaction to inhaled organic particles. Hypersensitivity pneumonitis is a clinical diagnosis and can be a challenging diagnosis to make. Both imaging and blood work can be variable and identification of the precipitating antigen is frequently never achieved. In fact, there is no consensus regarding diagnostic criteria for this disease (1, 2). Though there are no formal guidelines, multidisciplinary discussion and bronchoscopy with biopsy often play an important role in diagnosis, particularly when imaging is not typical for UIP.
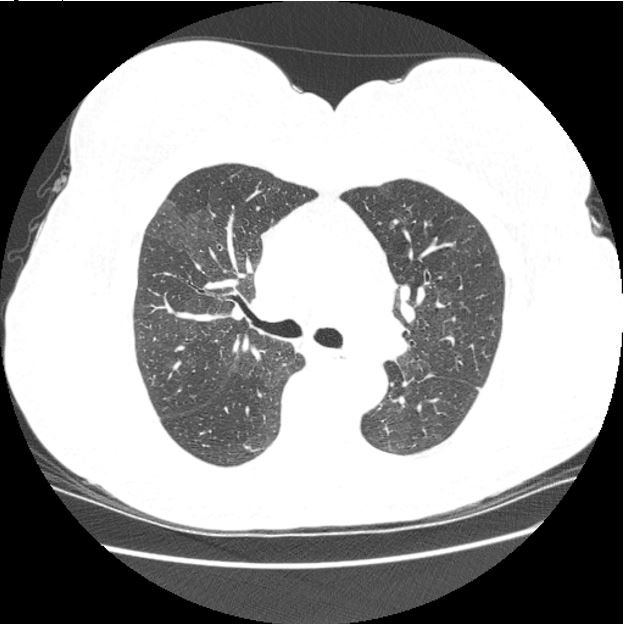
Hypersensitivity pneumonitis comes in three stages: acute, subacute, and chronic, all of which have a different set of symptoms and radiographic findings. Acute HP generally occurs a few hours after exposure and is characterized by flu-like symptoms including fever, cough, and wheezing. CT imaging may be normal or could demonstrate ground glass opacities consistent with pneumonitis. Subacute HP results from repeated exposures to low-level antigens. Patients usually present with insidious onset of malaise, dry cough, and dyspnea over a period of weeks to months. Imaging generally consists of ground glass opacities, diffuse micronodules, and mosaicism with areas of focal air-trapping (Figure 2). Chronic HP is the result of unrecognized/untreated acute and subacute episodes. Symptoms of dyspnea, cough and weight loss are common and imaging in advanced cases is often indistinguishable from IPF or fibrotic non-specific interstitial pneumonia (3).
Identification of HP is critical, as both treatment and prognosis are substantially different from IPF and other interstitial lung disease. The primary treatment modality is antigen avoidance, though this is not always feasible and an inciting antigen cannot be identified in over 50% of cases (4). The next step in treatment consists of immunosuppression, which begins with systemic corticosteroids for acute management. If there is response to corticosteroid, then the goal is to transition to a steroid-sparing medication such as azathioprine or leflunomide with the goal of slowing or preventing progression of disease. Immunosuppression is less likely to be helpful in advanced disease with fibrosis, and in these patients prognosis is quite poor and similar to that seen in individuals with IPF (5).
Figure 2:
References:
-
Riario Sforza GG, Marinou A. Hypersensitivity pneumonitis: a complex lung disease. Clin Mol Allergy 2017;15:6.
-
Lacasse Y, Girard M, Cormier Y. Recent advances in hypersensitivity pneumonitis. Chest 2012;142:208–217.
-
Selman M, Pardo A, King TE. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012;186:314–324.
-
Fernández Pérez ER, Swigris JJ, Forssén AV, Tourin O, Solomon JJ, Huie TJ, Olson AL, Brown KK. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest 2013;144:1644–1651.
-
Vourlekis JS, Schwarz MI, Cherniack RM, Curran-Everett D, Cool CD, Tuder RM, King TE, Brown KK. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004;116:662–668.


