Combing Through the Honeycomb
Sarah Kiel, MD, Jared Chiarchiaro, MD
Division of Pulmonary, Allergy, and Critical Care Medicine
University of Pittsburgh Medical Center, Pittsburgh, PA
Case
A previously healthy 74-year-old man presents with 2 years of progressive dyspnea on exertion and a persistent dry cough increasing in frequency and intensity over the same time period. He reports minimal tobacco use, 1 pack year accumulated 40 years prior. He reports no inhalational exposures through travel, environment, occupation, hobbies or pets. He mentions occasional thigh weakness and difficulty rising from a seated position, but denies any inflammatory arthralgias, rashes, Raynaud’s phenomena, sicca, reflux symptoms, or family history of autoimmune disease. Physical exam reveals a resting oxygen saturation of 92% on room air, no cyanosis, clubbing, or edema in extremities, and inspiratory crackles throughout the bases bilaterally. Recent serologic testing reveals no significant results from the following tests: antinuclear antibodies, myositis panel including anti-jo-1 antibody, anti-cyclic citrullinated peptide, rheumatoid factor, anti-Ro/SSA, anti-La/SSB antibodies. Pulmonary function tests (PFTs) demonstrate a restrictive pattern with reduced diffusing capacity for carbon monoxide. Representative CT images shown below.
Images (Fig 1A and 1B)
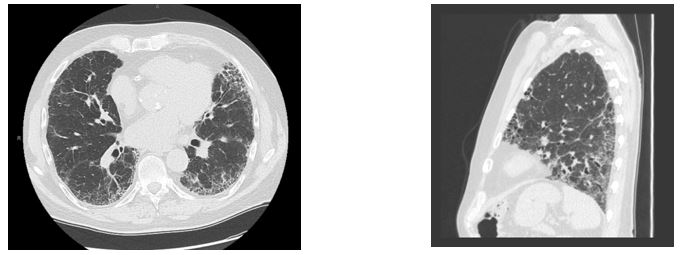
Question
Based on the information given, what further evaluation should be undertaken?
A. Bronchoscopy with transbronchial biopsies
B. Bronchoscopy with bronchoalveolar lavage (BAL)
C. VATS biopsy
D. No further evaluation is indicated
Answer: D. No further evaluation is indicated
Discussion
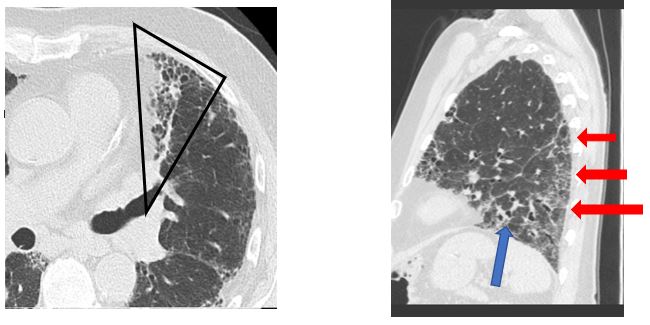
Traditionally, the diagnosis of idiopathic pulmonary fibrosis (IPF) is made via surgical lung biopsy. However, the clinical presentation and presence of specific radiographic findings are usually sufficient to make the diagnosis. High resolution CT (HRCT) images consistent with a UIP pattern of fibrosis are highly specific for the histopathologic correlate of IPF (1,2). The CT images depicted (Figs. 2A-B) demonstrate a typical UIP pattern characterized by: subpleural honeycombing (black outline), a predominantly basal distribution (red arrows), reticular pattern, traction bronchiectasis (blue arrows), and paucity of ground glass opacities (2). Most important is the paucity of ground glass opacities; their presence should prompt further evaluation and consideration for a diagnosis other than UIP (3). In the setting of a high pretest probability based on HRCT and clinical presentation, surgical lung biopsy is confirmatory but does not significantly change diagnosis or management and is associated with risks including procedural mortality (1.7%), respiratory infection (6.5%), and potential IPF exacerbation (6%) (3). As such, patients with a typical clinical presentation and a typical UIP pattern by HRCT, without identifiable exposure or other etiology, can be diagnosed with IPF without biopsy or BAL based on the 2018 ATS/ERS/JRS/ALAT clinical practice guidelines.
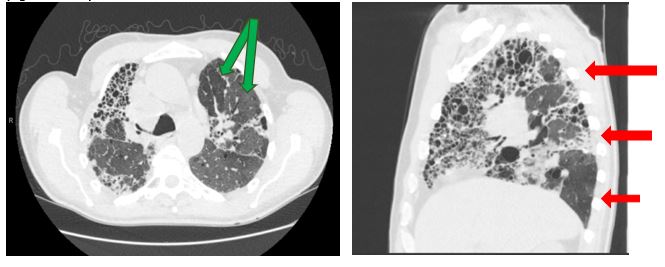
Given the risk associated with invasive diagnostic work up, it becomes imperative to recognize the differences between the four categories of HRCT imaging: typical for UIP, probable UIP, indeterminate for UIP, and features consistent with alternative diagnosis. Recommendation for more invasive work up would be indicated for the latter two categories. A probable UIP CT pattern remains the same as a typical UIP pattern in terms of a basal and subpleural heterogenous reticular distribution, and presence of peripheral traction bronchiectasis. However, honeycombing is often absent. In cases with a probable UIP pattern radiographically, 82-94% of cases will demonstrate a probable or definite UIP histopathological pattern on surgical lung biopsy (4). An indeterminate radiographic UIP pattern is often more diffuse in its interstitial markings without the expected basal and subpleural distribution. CT features that should prompt the pulmonologist to consider an alternative diagnosis to UIP include: upper lung or mid-lung fibrosis, subpleural sparing, any consolidation or extensive ground glass opacities, mosaicism (green arrows), and diffuse nodules or cysts (Figs. 3A-B). In multiple studies concerning fibrotic ILDs, the use of multidisciplinary evaluation with experienced clinicians, radiologists, and pathologist may change an ILD diagnosis in up to 20-50% of cases with an increase in diagnostic confidence (2). The two most common missed diagnoses for IPF by imaging are chronic hypersensitivity pneumonitis (HSP) and non-specific interstitial pneumonia (NSIP). Chronic HSP often demonstrates an upper to mid-lung distribution, mosaic attenuation or air trapping, and centrilobular nodules which aid in distinguishing it from a typical UIP pattern. NSIP often requires a surgical lung biopsy for diagnosis and radiographically may demonstrate subpleural sparing, a feature atypical for UIP.
(Fig 2A and 2B)
(Fig. 3A and 3B)
References:
-
Chung JH, Chawla A, Peljto AL, et al. CT scan findings of probable usual interstitial pneumonitis have a high predictive value for histologic usual interstitial pneumonitis. Chest 2015; 147(2): 450–9.
-
Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med 2018; 6(2): 138–53.
-
Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018;198(5): e44–e68.
-
Raghu G, Lynch D, Godwin JD, et al. Diagnosis of idiopathic pulmonary fibrosis with high-resolution CT in patients with little or no radiological evidence of honeycombing: secondary analysis of a randomised, controlled trial. Lancet Respir Med 2014; 2(4): 277–84.


